
The Hu lab is interested in studying molecular and cellular mechanisms involved in neuro-degeneration.
Frontotemporal Lobar Degeneration with ubiquitin and TDP-43 positive inclusions (FTLD-U) is a very common early onset dementia disease often associated with Amyotrophic Lateral Sclerosis (ALS). Mutations in Progranulin (PGRN), resulting in PGRN haplo-insufficiency, are one of the main causes of FTLD-TDP. A transmembrane protein of unknown function, TMEM106B, is a risk factor for FTLD-TDP with PGRN mutations. More recently, mutations in C9orf72, a gene of unknown function, were found to the main cause of mixed ALS/FTLD phenotypes. We are trying to understand the cellular functions of PGRN, TMEM106B and C9orf72 as well as other genes implicated in FTLD/ALS. We hope our studies will not only shed light on the molecular and cellular mechanisms of FTLD but also on the regulation of normal cellular functions by these FTLD proteins, such as regulation of autophagy-lysosome functions by PGRN, TMEM106B and C9orf72.
(Click figures for larger images – opens in new tab/window)
Project 1: Investigate signaling mechanisms of PGRN and its function in the lysosomes
Progranulin (PGRN) is a secreted glycoprotein of 7.5 granulin repeats. PGRN and granulin peptides have been implicated in many processes, including tumorigenesis, diabetes, wound healing, neuronal survival and inflammation. Elevated levels of PGRN have been associated with increased tumorigenesis and invasion. Mutations in PGRN, resulting in PGRN haplo-insufficiency, are a leading cause of frontotemporal lobar degeneration with ubiquitin positive inclusions (FTLD-U). Moreover, human patient with PGRN homozygous mutations develop neuronal ceroid lipofucinosis (NCL), a lysosome storage disorder, suggesting a role of PGRN in regulating lysosomal function. However, how PGRN regulates so many cellular processes are largely unknown. We are interested in the following aspect of PGRN biology:
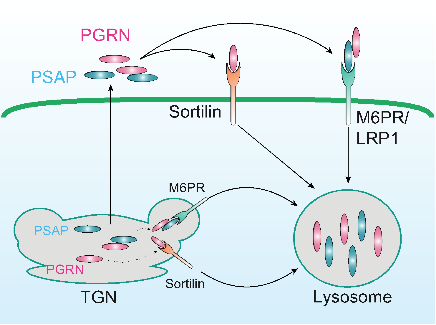
1.1 Determine the function of PGRN in the lysosome. We have shown a transmembrane protein of the VPS10 family, sortilin, mediates PGRN endocytosis and delivery into lysosomes (Neuron 68(4):654-67; PLoS One 2011;6(6):e21023). More recently, we uncovered a novel interaction between PGRN and prosaposin (PSAP) and demonstrate that prosaposin facilitates progranulin lysosomal trafficking via trafficking receptors M6PR/LRP1 independent of sortilin (J. Cell Bio., 2015) (Fig. 1). We are interested in further investigating the mechanisms controlling PGRN lysosomal trafficking and processing; the cross-regulation between PGRN and PSAP and other lysosomal functions of PGRN.
1.2 Determine signaling mechanisms of extracellular PGRN and granulin peptides. Extracellular PGRN and granulin peptides have been shown to regulate inflammation, neuronal survival and tumorigenesis. However, receptor and signaling pathways are still not clear. We are using proteomic and genomic approaches searching for PGRN signaling receptors.
(Click figures for larger images – opens in new tab/window)
Project 2: Investigate TMEM106B function and its interaction with PGRN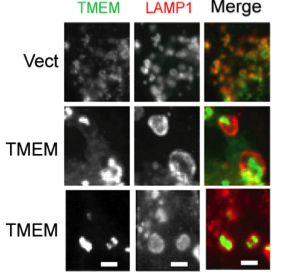
TMEM106B polymorphism affects the risk for FTLD, specifically in PGRN mutant carriers, suggesting a genetic connection between PGRN and TMEM106B. However, essentially nothing is known about the function and regulation of TMEM106B, which encodes a type II transmembrane protein. Our preliminary data demonstrated that TMEM106B mainly localizes in the late endosome/lysosome compartments and increased TMEM106B levels result in enlarged lysosomes (Fig. 2) and a delay in the degradation of endocytic cargoes. Furthermore, TMEM106B levels are modulated by lysosomal activities (Hum Mol Genet. 2013 Feb 15;22(4):685-95). We propose that TMEM106B and PGRN function together in the lysosomes to regulate lysosomal function.
2.1 Investigate TMEM106B functions. We are interested in further dissecting the role of TMEM106B and its homologs on lysosomal membrane using cell biological approaches.
2.2 Investigate mechanisms involved in regulating TMEM106B function. We found that TMEM106B is a substrate of regulated intramembrane proteolysis (RIP) (J Biol Chem. 2014 Jul 11;289(28):19670-19680). The importance of RIP in regulating TMEM106B function is currently under investigation.
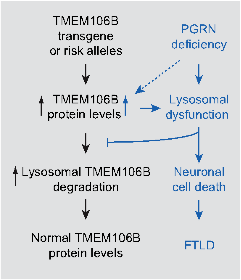
2.3 Determine the interaction between PGRN and TMEM106B. The TMEM106B polymorphism is a risk for FTLD with PGRN mutation, suggesting a genetic interaction between PGRN and TMEM106B. How PGRN and TMEM106B functionally interact is still not clear. In FTLD cases, increased TMEM106B levels are correlated with increased risk for FTLD with mutations in PGRN. We have generated transgenic mice overexpressing TMEM106B in the forebrain and found that TMEM106B protein levels are regulated by PGRN and increased TMEM106B levels exacerbates lysosomal abnormalities in PGRN deficient background. Our data led us to propose that PGRN deficiency leads to increased TMEM106B levels, which further exacerbates lysosomal dysfunction and neuronal cell death (Fig. 3). We are still dissecting cross-regulation between PGRN and TMEM106B.
(Click figures for larger images – opens in new tab/window)
Project 3: Investigate cellular functions of C9orf72

Hexanucleotide repeat expansion in the C9orf72 gene is a leading cause of frontotemporal lobar degeneration (FTLD) with amyotrophic lateral sclerosis (ALS). Reduced expression of C9orf72 has been proposed as a possible disease mechanism. However, the cellular function of C9orf72 is completely unknown. Through proteomic screen, we identified two binding partners of C9orf72, SMCR8 and WDR41. We show that WDR41 interacts with the C9orf72/SMCR8 heterodimer to form a ternary complex and WDR41 is tightly associated with the Golgi complex. We further demonstrate that C9orf72/SMCR8/WDR41 associates with the FIP200/ Ulk1 complex, which is essential for autophagy initiation (Fig. 4). C9orf72 deficient mice, generated using the CRISPR/Cas9 system, show severe splenomegaly. Increased levels of autophagy and lysosomal proteins were detected in both the spleen and liver of C9orf72 deficient mice, supporting an in vivo role of C9orf72 in regulating the autophagy/lysosome pathway.
3.1 Determine the role of C9orf72/SMCR8/WDR41 complex in autophagy. We plan to determine whether autophagy activation (such as Ulk1 activation, LC3 conversion) is affected by deletion of C9orf72, SMCR8 or WDR41. We will also investigate whether the C9orf72/SMCR8/WDR41 complex is a substrate for Ulk1 during autophagy activation.
3.2 Determine the role of the C9orf72/SMCR8/WDR41 complex in inflammation. Our data from C9orf72 deficient mice suggest a role of C9orf72 in regulating inflammation. We will further investigate the role of the C9orf72/SMCR8/WDR41 complex in macrophage activation and any potential regulation of this complex during inflammation.